ValspodarP-glycoprotein inhibitor CAS# 121584-18-7 |

- Dofequidar
Catalog No.:BCC4176
CAS No.:129716-58-1
- Elacridar
Catalog No.:BCC1546
CAS No.:143664-11-3
- Elacridar hydrochloride
Catalog No.:BCC1547
CAS No.:143851-98-3
- LY335979 (Zosuquidar 3HCL)
Catalog No.:BCC3878
CAS No.:167465-36-3
- Tariquidar
Catalog No.:BCC3625
CAS No.:206873-63-4
- Tariquidar methanesulfonate, hydrate
Catalog No.:BCC1986
CAS No.:625375-83-9
Quality Control & MSDS
Number of papers citing our products
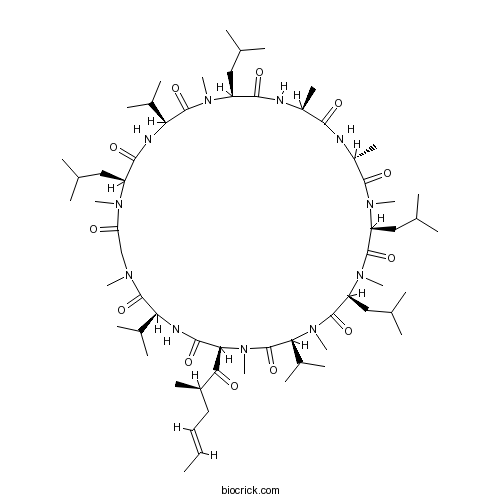
Chemical structure
3D structure
| Cas No. | 121584-18-7 | SDF | Download SDF |
| PubChem ID | 5281884 | Appearance | Powder |
| Formula | C63H111N11O12 | M.Wt | 1214.62 |
| Type of Compound | N/A | Storage | Desiccate at -20°C |
| Synonyms | Valspodar | ||
| Solubility | DMSO : 12 mg/mL (9.88 mM; Need ultrasonic and warming) H2O : < 0.1 mg/mL (insoluble) | ||
| Chemical Name | (3S,6S,9S,12R,15S,18S,21S,24S,30S,33S)-1,4,7,10,12,15,19,25,28-nonamethyl-33-[(E,2R)-2-methylhex-4-enoyl]-6,9,18,24-tetrakis(2-methylpropyl)-3,21,30-tri(propan-2-yl)-1,4,7,10,13,16,19,22,25,28,31-undecazacyclotritriacontane-2,5,8,11,14,17,20,23,26,29,32-undecone | ||
| SMILES | CC=CCC(C)C(=O)C1C(=O)NC(C(=O)N(CC(=O)N(C(C(=O)NC(C(=O)N(C(C(=O)NC(C(=O)NC(C(=O)N(C(C(=O)N(C(C(=O)N(C(C(=O)N1C)C(C)C)C)CC(C)C)C)CC(C)C)C)C)C)CC(C)C)C)C(C)C)CC(C)C)C)C)C(C)C | ||
| Standard InChIKey | YJDYDFNKCBANTM-QCWCSKBGSA-N | ||
| Standard InChI | InChI=1S/C63H111N11O12/c1-26-27-28-41(16)53(76)52-57(80)67-49(38(10)11)61(84)68(19)33-48(75)69(20)44(29-34(2)3)56(79)66-50(39(12)13)62(85)70(21)45(30-35(4)5)55(78)64-42(17)54(77)65-43(18)58(81)71(22)46(31-36(6)7)59(82)72(23)47(32-37(8)9)60(83)73(24)51(40(14)15)63(86)74(52)25/h26-27,34-47,49-52H,28-33H2,1-25H3,(H,64,78)(H,65,77)(H,66,79)(H,67,80)/b27-26+/t41-,42+,43-,44+,45+,46+,47+,49+,50+,51+,52+/m1/s1 | ||
| General tips | For obtaining a higher solubility , please warm the tube at 37 ℃ and shake it in the ultrasonic bath for a while.Stock solution can be stored below -20℃ for several months. We recommend that you prepare and use the solution on the same day. However, if the test schedule requires, the stock solutions can be prepared in advance, and the stock solution must be sealed and stored below -20℃. In general, the stock solution can be kept for several months. Before use, we recommend that you leave the vial at room temperature for at least an hour before opening it. |
||
| About Packaging | 1. The packaging of the product may be reversed during transportation, cause the high purity compounds to adhere to the neck or cap of the vial.Take the vail out of its packaging and shake gently until the compounds fall to the bottom of the vial. 2. For liquid products, please centrifuge at 500xg to gather the liquid to the bottom of the vial. 3. Try to avoid loss or contamination during the experiment. |
||
| Shipping Condition | Packaging according to customer requirements(5mg, 10mg, 20mg and more). Ship via FedEx, DHL, UPS, EMS or other couriers with RT, or blue ice upon request. | ||
| Description | P-glycoprotein (P-gp) modulator; inhibits P-gp-mediated multidrug-resistance (MDR). Reverses resistance to several cytotoxic drugs including mitoxantrone and doxorubicin (resistance factors are 2.0 and 6.5 respectively) in human MDR cancer cell lines. Non-immunosuppressive analog of cyclosporin A. |

Valspodar Dilution Calculator
Valspodar Molarity Calculator
| 1 mg | 5 mg | 10 mg | 20 mg | 25 mg | |
| 1 mM | 0.8233 mL | 4.1165 mL | 8.233 mL | 16.4661 mL | 20.5826 mL |
| 5 mM | 0.1647 mL | 0.8233 mL | 1.6466 mL | 3.2932 mL | 4.1165 mL |
| 10 mM | 0.0823 mL | 0.4117 mL | 0.8233 mL | 1.6466 mL | 2.0583 mL |
| 50 mM | 0.0165 mL | 0.0823 mL | 0.1647 mL | 0.3293 mL | 0.4117 mL |
| 100 mM | 0.0082 mL | 0.0412 mL | 0.0823 mL | 0.1647 mL | 0.2058 mL |
| * Note: If you are in the process of experiment, it's necessary to make the dilution ratios of the samples. The dilution data above is only for reference. Normally, it's can get a better solubility within lower of Concentrations. | |||||

Calcutta University

University of Minnesota

University of Maryland School of Medicine

University of Illinois at Chicago

The Ohio State University

University of Zurich

Harvard University

Colorado State University

Auburn University

Yale University

Worcester Polytechnic Institute

Washington State University

Stanford University

University of Leipzig

Universidade da Beira Interior

The Institute of Cancer Research

Heidelberg University

University of Amsterdam

University of Auckland

TsingHua University

The University of Michigan

Miami University

DRURY University

Jilin University

Fudan University

Wuhan University

Sun Yat-sen University

Universite de Paris

Deemed University

Auckland University

The University of Tokyo

Korea University
Valspodar is a potent inhibitor of P-glycoprotein (P-gp) widely used in preclinical and clinical studies [1].
P-gp is a transmembrane glycoprotein which is located on cell membrane. P-gp distributes extensively and is expressed in certain cell types primarily containing liver, colon, kidney and pancreas. It also is known as multidrug resistance protein 1 (MDR1) which is pumps foreign substances out of cells. P-gp decreases the net uptake of cytotoxic drugs into the cells and mediats the efflux of these agents out of the cells, which is ATP-dependent. P-gp also overexpress in some cancer cells. P-gp plays an important role in mediating resistance to anticancer drugs and decreasing drug accumulation in multidrug-resistant cancer cells.[1]
Valspodar can reverse the resistance to mitoxantrane which is due to the expression of P-gp. The IC50 of mitoxantrane decreased from 1.6 ± 0.13 μM to 0.4 ± 0.02
μM in MDA-MB-435mdr cells pretreated with 3 mg/ml PSC. Valspodar increase the mitoxantrane intracellular accumulation by decreasing drug efflux and increasing mitoxantrone net uptake in cells.[1] The cytotoxicity was significant greater in T47D/TAMR-6 cells treated with doxorubicin and valspodar than doxorubicin only. Co-encapsulation of doxorubicin and valspodar presents a promising anticancer effect.[2] Valspodar was rapid absorpted and reachs the peak within 2 hnafter an oral dose. Valspodar showed properties of wide distribution, low hepatic extraction and mean bioavailability of 42.8% in rat.[3]
References:
[1]. Shen F, Bailey BJ, Chu S, Bence AK, Xue X, Erickson P, Safa AR, Beck WT, Erickson LC: Dynamic assessment of mitoxantrone resistance and modulation of multidrug resistance by valspodar (PSC833) in multidrug resistance human cancer cells. J Pharmacol Exp Ther 2009, 330(2):423-429.
[2]. Bajelan E, Haeri A, Vali AM, Ostad SN, Dadashzadeh S: Co-delivery of doxorubicin and PSC 833 (Valspodar) by stealth nanoliposomes for efficient overcoming of multidrug resistance. J Pharm Pharm Sci 2012, 15(4):568-582.
[3]. Binkhathlan Z, Hamdy DA, Brocks DR, Lavasanifar A: Pharmacokinetics of PSC 833 (valspodar) in its Cremophor EL formulation in rat. Xenobiotica 2010, 40(1):55-61.
- DMCM hydrochloride
Catalog No.:BCC7560
CAS No.:1215833-62-7
- Gatifloxacin hydrochloride
Catalog No.:BCC4224
CAS No.:121577-32-0
- NBI 27914 hydrochloride
Catalog No.:BCC7124
CAS No.:1215766-76-9
- CP-809101 hydrochloride
Catalog No.:BCC1499
CAS No.:1215721-40-6
- RS 100329 hydrochloride
Catalog No.:BCC5741
CAS No.:1215654-26-4
- SB 242084
Catalog No.:BCC5949
CAS No.:1215566-78-1
- CFM 1571 hydrochloride
Catalog No.:BCC5924
CAS No.:1215548-30-3
- RG2833
Catalog No.:BCC1893
CAS No.:1215493-56-3
- GR 144053 trihydrochloride
Catalog No.:BCC6998
CAS No.:1215333-48-4
- SR 58611A hydrochloride
Catalog No.:BCC7833
CAS No.:121524-09-2
- Salvianolic acid B; Lithospermic acid B; Danfensuan B
Catalog No.:BCC8249
CAS No.:121521-90-2
- PF-04991532
Catalog No.:BCC8094
CAS No.:1215197-37-7
- 6-Demethoxy-9'-deoxycleomiscosin A
Catalog No.:BCN7298
CAS No.:121587-18-6
- 6-Demethoxycleomiscosin A
Catalog No.:BCN7299
CAS No.:121587-20-0
- YM 298198 hydrochloride
Catalog No.:BCC7366
CAS No.:1216398-09-2
- SB 258585 hydrochloride
Catalog No.:BCC7216
CAS No.:1216468-02-8
- Kaempferol-3-O-(2',6'-di-O-trans-p-coumaroyl)-beta-D-glucopyranoside
Catalog No.:BCN1603
CAS No.:121651-61-4
- BX 513 hydrochloride
Catalog No.:BCC5940
CAS No.:1216540-18-9
- ZK 93423 hydrochloride
Catalog No.:BCC7227
CAS No.:1216574-52-5
- 2-Cyclopropyl-4-(4-fluorophenyl)-quinolyl-3-methanol
Catalog No.:BCC8574
CAS No.:121660-11-5
- 2-Cyclopropyl-4-(4-fluorophenyl)quinoline-3-carboxaldehyde
Catalog No.:BCC8573
CAS No.:121660-37-5
- Trap 101
Catalog No.:BCC7390
CAS No.:1216621-00-9
- SCH 79797 dihydrochloride
Catalog No.:BCC7125
CAS No.:1216720-69-2
- BYK 191023 dihydrochloride
Catalog No.:BCC7506
CAS No.:1216722-25-6
P-glycoprotein modulation by valspodar and cyclosporin does not increase tumor uptake of doxorubicin administered via isolated lung perfusion to rats bearing sarcoma lung metastases.[Pubmed:21737631]
Anticancer Res. 2011 Jun;31(6):2121-8.
BACKGROUND: Isolated lung perfusion (ILP) with doxorubicin allows a regional increase in drug exposure while sparing unaffected tissues, but clinical results have so far been disappointing, presumably in part because of the limited tumor penetration of doxorubicin. The aim of this study was to assess whether tumor uptake of doxorubicin, administered locoregionally by ILP, would be increased by the administration of P-glycoprotein (P-gp) modulators. MATERIALS AND METHODS: Single-pass antegrade ILP (A-ILP) was performed with doxorubicin in rats bearing a pulmonary sarcoma nodule which were either untreated or received P-gp inhibitors cyclosporin, Valspodar or the vehicle, Cremophor(R), only. Doxorubicin concentrations in tumor, lung and effluent were measured by high performance liquid chromatography (HPLC) coupled to spectrofluorimetric detection and the expression of P-gp was examined by Western blot in tumors and lungs. RESULTS: Doxorubicin concentrations in tumors were 5- to 10-fold lower than those measured in lungs tissues. Doxorubicin penetration in tumors, expressed as tumor retention ratios (TR60min), were not different between the groups. Western blot analysis did not show any evidence of baseline or doxorubicin-induced P-gp expression in the tumor model. CONCLUSION: P-gp modulation with cyclosporin or Valspodar fails to increase the tumor uptake of doxorubin administered by A-ILP. Other reasons for low doxorubicin penetration in tumor, such as high interstitial fluid pressure or tumor vasculature barrier, or alternate cell membrane drug transporters, need to be examined for a better understanding of impaired doxorubicin delivery to tumor.
Characterization of the self assembly of methoxy poly(ethylene oxide)-block-poly(alpha-benzyl carboxylate-epsilon-caprolactone) for the solubilization and in vivo delivery of valspodar.[Pubmed:22283648]
Curr Drug Deliv. 2012 Mar;9(2):164-71.
The aim of this study was to characterize the nanostructures formed from assembly of poly(ethylene oxide)-bpoly( alpha-benzyl carboxylate epsilon-caprolactone) (PEO-b-PBCL) in water, determine the effect of weight fraction of the hydrophilic block( fEO) on their morphology, and to investigate their potential for solubilization and delivery of P-glycoprotein (P-gp) inhibitor, Valspodar. Three PEO-b-PBCL block copolymers having fEO ranging from 0.18-0.40 were synthesized. Assembly of PEO-b-PBCL was triggered through a co-solvent evaporation method. The average critical aggregation concentration (CAC) for PEO114-b-PBCL(3)(0), PEO(1)(1)(4)-b-PBCL(6)(0), and PEO(1)(1)(4)-b-PBCL(9)(5) was found to be 62, 41, and 23 nM, respectively. A lower rigidity of the hydrophobic domain in nanostructures formed from the assembly of PEO(1)(1)(4)-b- PBCL(6)(0) and PEO(1)(1)(4)-b-PBCL(9)(5) in comparison to PEO(1)(1)(4)-b-PBCL(3)(0) was observed. The morphology of the assembled structures was characterized by transmission electron microscopy (TEM). The TEM images of PEO(1)(1)(4)-b-PBCL(3)(0) (fEO = 0.40) showed the formation of spherical micelles with high polydispersity, whereas the assembly of PEO(1)(1)(4)-b-PBCL(6)(0) (fEO = 0.25) and PEO(1)(1)(4)-b-PBCL95 (fEO = 0.18) resulted in a mixed population of spherical micelles and vesicles. Valspodar achieved high loading in all the three PEO-b-PBCL nanocarriers reaching aqueous solubility of nearly 2 mg/mL. The morphology of PEO-b-PBCL carrier did not seem to influence the pharmacokinetics of the encapsulated Valspodar in rats following intravenous administration. In conclusion, the results show a potential for PEO-b-PBCL nanocarriers as efficient solubilizing agents for delivery of Valspodar.
A double blinded, placebo-controlled pilot study to examine reduction of CD34 (+)/CD117 (+)/CD133 (+) lymphoma progenitor cells and duration of remission induced by neoadjuvant valspodar in dogs with large B-cell lymphoma.[Pubmed:28357033]
F1000Res. 2015 Feb 11;4:42.
We previously described a population of lymphoid progenitor cells (LPCs) in canine B-cell lymphoma defined by retention of the early progenitor markers CD34 and CD117 and "slow proliferation" molecular signatures that persist in the xenotransplantation setting. We examined whether Valspodar, a selective inhibitor of the ATP binding cassette B1 transporter (ABCB1, a.k.a., p-glycoprotein/multidrug resistance protein-1) used in the neoadjuvant setting would sensitize LPCs to doxorubicin and extend the length of remission in dogs with therapy naive large B-cell lymphoma. Twenty dogs were enrolled into a double-blinded, placebo controlled study where experimental and control groups received oral Valspodar (7.5 mg/kg) or placebo, respectively, twice daily for five days followed by five treatments with doxorubicin 21 days apart with a reduction in the first dose to mitigate the potential side effects of ABCB1 inhibition. Lymph node and blood LPCs were quantified at diagnosis, on the fourth day of neoadjuvant period, and 1-week after the first chemotherapy dose. Valspodar therapy was well tolerated. There were no differences between groups in total LPCs in lymph nodes or peripheral blood, nor in event-free survival or overall survival. Overall, we conclude that Valspodar can be administered safely in the neoadjuvant setting for canine B-cell lymphoma; however, its use to attenuate ABCB1 (+) cells does not alter the composition of lymph node or blood LPCs, and it does not appear to be sufficient to prolong doxorubicin-dependent remissions in this setting.
Co-delivery of doxorubicin and PSC 833 (Valspodar) by stealth nanoliposomes for efficient overcoming of multidrug resistance.[Pubmed:23106959]
J Pharm Pharm Sci. 2012;15(4):568-82.
PURPOSE: This study was aimed at developing co-encapsulated stealth nanoliposomes containing PSC 833, an efficient MDR modulator, and doxorubicin (DOX) in order to increase the effectiveness and decrease adverse effects of the anticancer drug. METHODS: In attempt to increase the encapsulation efficiency of drugs, different methods for liposome preparation were tested and the effect of different parameters such as drug to lipid molar ratio, cholesterol mole percent and lipid compositions, were investigated. The final product with a lipid composition of EPC:DSPE-PEG2000:Chol (60:5:30 %mol) was prepared by thin layer film hydration method. After preparation of empty liposomes, DOX and PSC 833 were loaded using ammonium sulfate gradient and remote film loading methods, respectively. Physical characteristics of optimized liposomes (DOX/PSC-L) such as particle size, zeta potential, encapsulation efficiency, in-vitro drugs release and stability were evaluated. Furthermore, in vitro cytotoxicity study of various liposomal formulations as well as drugs, solutions against resistant human breast cancer cell line, T47D/TAMR-6, was evaluated using MTT assay. RESULTS: The best formulation showed a narrow size distribution with average diameter of 91.3 +/- 0.2 nm with zeta potential of -6 +/- 1.2, the encapsulation efficiency for DOX and PSC 833 were more than 95% and 65.5%, respectively. In DOX-resistant T47D/TAMR-6 cells, dual-agent stealth liposomes showed significantly greater cytotoxicity (P < 0.05) than free DOX and liposomal DOX plus free PSC 833 treatments. CONCLUSIONS: Co-encapsulation of DOX and PSC 833 presents a promising anticancer formulation, capable of effective reversal of drug resistance, and should be explored further in therapeutic studies with animal tumor xenograft models. This article is open to POST-PUBLICATION REVIEW. Registered readers (see "For Readers") may comment by clicking on ABSTRACT on the issue's contents page.
Dynamic assessment of mitoxantrone resistance and modulation of multidrug resistance by valspodar (PSC833) in multidrug resistance human cancer cells.[Pubmed:19423841]
J Pharmacol Exp Ther. 2009 Aug;330(2):423-9.
P-glycoprotein (Pgp), a member of the ATP-binding cassette transporter family, is one of the major causes for multidrug resistance (MDR). We report using confocal microscopy to study the roles of Pgp in mediating the efflux of the anticancer agent mitoxantrone and the reversal of MDR by the specific Pgp inhibitor Valspodar (PSC833). The net uptake and efflux of mitoxantrone and the effect of PSC833 were quantified and compared in Pgp-expressing human cancer MDA-MB-435 (MDR) cells and in parental wild-type cells. The MDR cells, transduced with the human Pgp-encoding gene MDR1 construct, were approximately 8-fold more resistant to mitoxantrone than the wild-type cells. Mitoxantrone accumulation in the MDR cells was 3-fold lower than that in the wild-type cells. The net uptake of mitoxantrone in the nuclei and cytoplasm of MDR cells was only 58 and 67% of that in the same intracellular compartment of the wild-type cells. Pretreatment with PSC833 increased the accumulation of mitoxantrone in the MDR cells to 85% of that in the wild-type cells. In living animals, the accumulation of mitoxantrone in MDA-MB-435mdr xenograft tumors was 61% of that in the wild-type tumors. Administration of PSC833 to animals before mitoxantrone treatment increased the accumulation of mitoxantrone in the MDR tumors to 94% of that in the wild-type tumors. These studies have added direct in vitro and in vivo visual information on how Pgp processes anticancer compounds and how Pgp inhibitors modulate MDR in resistant cancer cells.
Quantitation of doxorubicin uptake, efflux, and modulation of multidrug resistance (MDR) in MDR human cancer cells.[Pubmed:17947497]
J Pharmacol Exp Ther. 2008 Jan;324(1):95-102.
P-glycoprotein (Pgp), a membrane transporter encoded by the MDR1 gene in human cells, mediates drug efflux from cells, and it plays a major role in causing multidrug resistance (MDR). Confocal microscopy was used to study in vitro and in vivo drug accumulation, net uptake and efflux, and MDR modulation by P-glycoprotein inhibitors in MDR1-transduced human MDA-MB-435mdr (MDR) cancer cells. The MDR cells were approximately 9-fold more resistant to the anticancer drug doxorubicin than their parental wild-type MDA-MB-435wt (WT) cells. Doxorubicin accumulation in the MDR cells was only 19% of that in the WT cells. The net uptake of doxorubicin in the nuclei of the MDR cells was 2-fold lower than that in the nuclei of the WT cells. Pgp inhibitors verapamil, cyclosporine A, or PSC833 increased doxorubicin accumulation in the MDR cells up to 79%, and it reversed drug resistance in these cells. In living animals, doxorubicin accumulation in MDA-MB-435mdr xenograft tumors was 68% of that in the wild-type tumors. Administration of verapamil, cyclosporine A, or PSC833 before doxorubicin treatment of the animals increased doxorubicin accumulation in the MDR tumors up to 94%. These studies have added direct in vitro and in vivo information on the capacity of the transporter protein Pgp to efflux doxorubicin and on the reversal of MDR by Pgp inhibitors in resistant cancer cells.
Complete inhibition of P-glycoprotein by simultaneous treatment with a distinct class of modulators and the UIC2 monoclonal antibody.[Pubmed:17050779]
J Pharmacol Exp Ther. 2007 Jan;320(1):81-8.
P-glycoprotein (Pgp) is one of the active efflux pumps that are able to extrude a large variety of chemotherapeutic drugs from the cells, causing multidrug resistance. The conformation-sensitive UIC2 monoclonal antibody potentially inhibits Pgp-mediated substrate transport. However, this inhibition is usually partial, and its extent is variable because UIC2 binds only to 10 to 40% Pgp present in the cell membrane. The rest of the Pgp molecules become recognized by this antibody only in the presence of certain substrates or modulators, including vinblastine, cyclosporine A (CsA), and SDZ PSC 833 (Valspodar). Simultaneous application of any of these modulators and UIC2, followed by the removal of the modulator, results in a completely restored steady-state accumulation of various Pgp substrates (calcein-AM, daunorubicin, and 99mTc-hexakis-2-methoxybutylisonitrile), indicating near 100% inhibition of pump activity. Remarkably, the inhibitory binding of the antibody is brought about by coincubation with concentrations of CsA or SDZ PSC 833 approximately 20 times lower than what is necessary for Pgp inhibition when the modulators are applied alone. The feasibility of such a combinative treatment for in vivo multidrug resistance reversal was substantiated by the dramatic increase of daunorubicin accumulation in xenotransplanted Pgp+ tumors in response to a combined treatment with UIC2 and CsA, both administered at doses ineffective when applied alone. These observations establish the combined application of a class of modulators used at low concentrations and of the UIC2 antibody as a novel, specific, and effective way of blocking Pgp function in vivo.
Modulation of the tumor disposition of vinca alkaloids by PSC 833 in vitro and in vivo.[Pubmed:9864280]
J Pharmacol Exp Ther. 1998 Dec;287(3):963-8.
PSC 833, a nonimmunosuppressive cyclosporin, is able to inhibit the efflux of antitumor drugs mediated by P-glycoprotein (P-gp). The purpose of the present study is to compare the effect of PSC 833 on the tumor disposition of [3H]vincristine ([3H]VCR) and [3H]vinblastine ([3H]VBL) in in vitro and in vivo experiments from a pharmacokinetic point of view. In in vitro experiments, the effect of PSC 833 was investigated on the cellular uptake of [3H]VCR and [3H]VBL by HCT-15 and COLO 205, human colorectal tumor cell lines with extensive and minimal expression of P-gp, respectively. PSC 833 (2 microM) increased the cellular uptake of [3H]VCR and [3H]VBL by HCT-15 cells, but not that by COLO 205 cells, 8- and 6-fold, respectively, without affecting the initial influx rates. In addition, 2 microM PSC 833 reduced the efflux of [3H]VCR from HCT-15 cells to a level comparable with that from COLO 205 cells. Furthermore, the effect of PSC 833 on the tumor disposition of intravenously administered [3H]VCR and [3H]VBL was studied in tumor inoculated mice. Infusion of PSC 833 (10 microg/hr/mouse) increased the HCT-15 tumor disposition of [3H]VBL and [3H]VCR in vivo to a level comparable with that observed in vitro. These findings demonstrate that PSC 833 enhances the tumor disposition of vinca alkaloids by inhibition of P-gp-mediated efflux not only in vitro but also in vivo in a solid tumor model.
