Nelfinavir MesylateHIV protease inhibitor,antiretroviral drug for HIV treatment CAS# 159989-65-8 |

- Daptomycin
Catalog No.:BCC1057
CAS No.:103060-53-3
- Gemcitabine HCl
Catalog No.:BCC1076
CAS No.:122111-03-9
- Clofarabine
Catalog No.:BCC1078
CAS No.:123318-82-1
- Ifosfamide
Catalog No.:BCC1164
CAS No.:3778-73-2
- Carboplatin
Catalog No.:BCC1170
CAS No.:41575-94-4
Quality Control & MSDS
Number of papers citing our products
Chemical structure
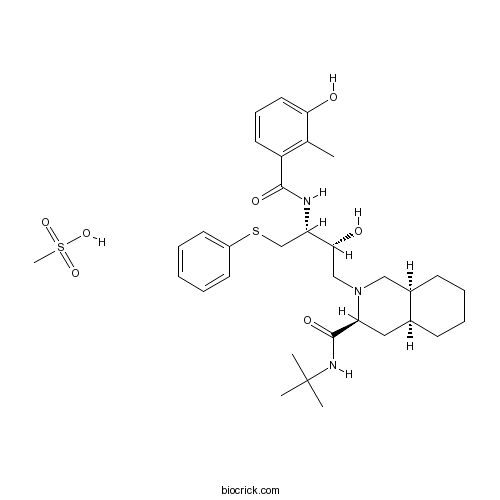
3D structure
| Cas No. | 159989-65-8 | SDF | Download SDF |
| PubChem ID | 64142 | Appearance | Powder |
| Formula | C33H49N3O7S2 | M.Wt | 663.89 |
| Type of Compound | N/A | Storage | Desiccate at -20°C |
| Synonyms | AG 1341 | ||
| Solubility | DMSO : ≥ 25 mg/mL (37.66 mM) *"≥" means soluble, but saturation unknown. | ||
| Chemical Name | (3S,4aS,8aS)-N-tert-butyl-2-[(2R,3R)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylsulfanylbutyl]-3,4,4a,5,6,7,8,8a-octahydro-1H-isoquinoline-3-carboxamide;methanesulfonic acid | ||
| SMILES | CC1=C(C=CC=C1O)C(=O)NC(CSC2=CC=CC=C2)C(CN3CC4CCCCC4CC3C(=O)NC(C)(C)C)O.CS(=O)(=O)O | ||
| Standard InChIKey | NQHXCOAXSHGTIA-SKXNDZRYSA-N | ||
| Standard InChI | InChI=1S/C32H45N3O4S.CH4O3S/c1-21-25(15-10-16-28(21)36)30(38)33-26(20-40-24-13-6-5-7-14-24)29(37)19-35-18-23-12-9-8-11-22(23)17-27(35)31(39)34-32(2,3)4;1-5(2,3)4/h5-7,10,13-16,22-23,26-27,29,36-37H,8-9,11-12,17-20H2,1-4H3,(H,33,38)(H,34,39);1H3,(H,2,3,4)/t22-,23+,26-,27-,29+;/m0./s1 | ||
| General tips | For obtaining a higher solubility , please warm the tube at 37 ℃ and shake it in the ultrasonic bath for a while.Stock solution can be stored below -20℃ for several months. We recommend that you prepare and use the solution on the same day. However, if the test schedule requires, the stock solutions can be prepared in advance, and the stock solution must be sealed and stored below -20℃. In general, the stock solution can be kept for several months. Before use, we recommend that you leave the vial at room temperature for at least an hour before opening it. |
||
| About Packaging | 1. The packaging of the product may be reversed during transportation, cause the high purity compounds to adhere to the neck or cap of the vial.Take the vail out of its packaging and shake gently until the compounds fall to the bottom of the vial. 2. For liquid products, please centrifuge at 500xg to gather the liquid to the bottom of the vial. 3. Try to avoid loss or contamination during the experiment. |
||
| Shipping Condition | Packaging according to customer requirements(5mg, 10mg, 20mg and more). Ship via FedEx, DHL, UPS, EMS or other couriers with RT, or blue ice upon request. | ||
| Description | Orally active human immunodeficiency virus protease inhibitor. Potently inhibits HIV-1 protease (Ki = 2 nM) in vitro. |

Nelfinavir Mesylate Dilution Calculator
Nelfinavir Mesylate Molarity Calculator
| 1 mg | 5 mg | 10 mg | 20 mg | 25 mg | |
| 1 mM | 1.5063 mL | 7.5314 mL | 15.0627 mL | 30.1255 mL | 37.6568 mL |
| 5 mM | 0.3013 mL | 1.5063 mL | 3.0125 mL | 6.0251 mL | 7.5314 mL |
| 10 mM | 0.1506 mL | 0.7531 mL | 1.5063 mL | 3.0125 mL | 3.7657 mL |
| 50 mM | 0.0301 mL | 0.1506 mL | 0.3013 mL | 0.6025 mL | 0.7531 mL |
| 100 mM | 0.0151 mL | 0.0753 mL | 0.1506 mL | 0.3013 mL | 0.3766 mL |
| * Note: If you are in the process of experiment, it's necessary to make the dilution ratios of the samples. The dilution data above is only for reference. Normally, it's can get a better solubility within lower of Concentrations. | |||||

Calcutta University

University of Minnesota

University of Maryland School of Medicine

University of Illinois at Chicago

The Ohio State University

University of Zurich

Harvard University

Colorado State University

Auburn University

Yale University

Worcester Polytechnic Institute

Washington State University

Stanford University

University of Leipzig

Universidade da Beira Interior

The Institute of Cancer Research

Heidelberg University

University of Amsterdam

University of Auckland

TsingHua University

The University of Michigan

Miami University

DRURY University

Jilin University

Fudan University

Wuhan University

Sun Yat-sen University

Universite de Paris

Deemed University

Auckland University

The University of Tokyo

Korea University
Nelfinavir Mesylate is a potent inhibitor of HIV-1 protease [1].
HIV-1 protease is a constitutive enzyme that processes gag and gag-pol polyproteins for packaging into the nascent virion actively budding from a productively infected cell. Inhibition of the enzyme results in the formation of immature non-infectious particles [2].
Nelfinavir Mesylate is a potent inhibitor of HIV-1 protease with Ki of 2.0 nM. In CEM cells infected with the HIV strain IIIB, Nelfinavir Mesylate was potent antiviral agent with ED50 value of 14 nM and exhibited minimal cellular toxicity (TD50s > 5000 nM) [1]. In CEM-SS and MT-2 cells, Nelfinavir Mesylate protected these cells from acute HIV-1 RF- and HIV-1 IIIB-induced cell killing with EC50s ranging from 31 to 43 nM [3].
In 65 HIV-1-infected patients, Nelfinavir Mesylate was well-tolerated and exhibited robust antiviral activity with demonstrable superiority of the 750 mg and 1000 mg three times daily regimens. Thirty patients who continued to receive therapy at 12 months acquired a persistent 1.6 log10 reduction in HIV RNA, accompanied by a mean increase in CD4 cells of 180-200/mm3 [2].
References:
[1]. Kaldor SW, Kalish VJ, Davies JF, et al. Viracept (nelfinavir mesylate, AG1343): a potent, orally bioavailable inhibitor of HIV-1 protease. J Med Chem, 1997, 40(24): 3979-3985.
[2]. Markowitz M, Conant M, Hurley A, et al. A preliminary evaluation of nelfinavir mesylate, an inhibitor of human immunodeficiency virus (HIV)-1 protease, to treat HIV infection. J Infect Dis, 1998, 177(6): 1533-1540.
[3]. Patick AK, Mo H, Markowitz M, et al. Antiviral and resistance studies of AG1343, an orally bioavailable inhibitor of human immunodeficiency virus protease. Antimicrob Agents Chemother, 1996, 40(2): 292-297.
- Nelfinavir
Catalog No.:BCC4138
CAS No.:159989-64-7
- NIBR189
Catalog No.:BCC8056
CAS No.:1599432-08-2
- 3,5-Di-O-caffeoylquinic acid methyl ester
Catalog No.:BCN6491
CAS No.:159934-13-1
- Mc-Val-Cit-PABC-PNP
Catalog No.:BCC4028
CAS No.:159857-81-5
- 4'-O-Methylirenolone
Catalog No.:BCN7174
CAS No.:159853-36-8
- Sikokianin C
Catalog No.:BCN6827
CAS No.:159813-69-1
- Ulipristal
Catalog No.:BCC4944
CAS No.:159811-51-5
- Fmoc-Lys(Ac)-OH
Catalog No.:BCC3514
CAS No.:159766-56-0
- Ibutamoren Mesylate
Catalog No.:BCC1638
CAS No.:159752-10-0
- ISRIB (trans-isomer)
Catalog No.:BCC5340
CAS No.:1597403-47-8
- Wikstrol A
Catalog No.:BCN7938
CAS No.:159736-35-3
- Saropyrone
Catalog No.:BCN7692
CAS No.:159650-12-1
- Iniparib (BSI-201)
Catalog No.:BCC2208
CAS No.:160003-66-7
- Antibiotic AB 4063B
Catalog No.:BCN1827
CAS No.:160041-33-8
- Sambutoxin
Catalog No.:BCN1709
CAS No.:160047-56-3
- Huwentoxin XVI
Catalog No.:BCC8041
CAS No.:1600543-88-1
- Cryptofolione
Catalog No.:BCN7197
CAS No.:160098-78-2
- SCH 58261
Catalog No.:BCC7306
CAS No.:160098-96-4
- 2-Iminopiperidine hydrochloride
Catalog No.:BCC6862
CAS No.:16011-96-4
- L-BMAA hydrochloride
Catalog No.:BCC7400
CAS No.:16012-55-8
- 7-Chloro-1,2,3,4-tetrahydrobenzo[b]azepin-5-one
Catalog No.:BCC8779
CAS No.:160129-45-3
- BIM 23127
Catalog No.:BCC5822
CAS No.:160161-61-5
- SR 11302
Catalog No.:BCC3607
CAS No.:160162-42-5
- 14-Deoxy-11-hydroxyandrographolide
Catalog No.:BCN4702
CAS No.:160242-09-1
Quality-by-design based development of a self-microemulsifying drug delivery system to reduce the effect of food on Nelfinavir mesylate.[Pubmed:26854426]
Int J Pharm. 2016 Mar 30;501(1-2):311-25.
Poor aqueous solubility and moderate permeability of Nelfinavir Mesylate (NFM) leads to high variability in absorption after oral administration. To improve the solubility and bioavailability of NFM, the self microemulsifying drug delivery system (SMEDDS) was developed. For this purpose, Quality by design (QbD) approach employing D-optimal mixture design was used to prepare SMEDDS of NFM. Further, the software generated numerically optimized SMEDDS were developed by utilizing desirability function. Maisine 35-1, Tween 80, and Transcutol HP were identified as oil, surfactant, and co-surfactant that had best solubility for NFM. Ternary phase diagrams were plotted to identify the self-emulsification region. Dissolution of putative NFM in simulated fasted or fed small intestinal conditions, respectively, predicted that there is a positive food effect. However, NFM loaded SMEDDS showed absence of food effect with no significant difference in dissolution performance either in Fasted or fed state simulated intestinal fluid (FaSSIF or FeSSIF) biorelevent dissolution media. The prepared SMEDDS were thermodynamically stable with droplet size (121 nm), poly dispersity index (PDI) (0.198) and emulsification time (<1 min). Transmission electron microscopy (TEM) analysis confirmed the spherical shape of the reconstituted SMEDDS droplets. The ex vivo performance revealed 4.57 fold enhancement in the apparent permeability of NFM as compared to NFM suspension. The animal pharmacokinetic analysis in New Zealand strain rabbits indicated food effect on pure NFM suspension. However, absence of food effect and 3.5-3.6 fold enhancement in the oral bioavailability was observed when NFM was formulated into SMEDDS. Thus, it could be envisaged that development of SMEDDS formulation of NFM could be one of the best alternative to enhance oral bioavailability of NFM.
Novel nelfinavir mesylate loaded d-alpha-tocopheryl polyethylene glycol 1000 succinate micelles for enhanced pediatric anti HIV therapy: In vitro characterization and in vivo evaluation.[Pubmed:25270729]
Colloids Surf B Biointerfaces. 2014 Nov 1;123:302-10.
Worldwide more than 35 million people are living with Human Immunodeficiency Virus (HIV) where 3.3 million are children. This translates in approximately 700 new daily infections in children only in 2012. Prolonged High Activity Antiretroviral Therapy (HAART) regimes could present low-patient compliance, especially in children, affecting therapeutic success. Nelfinavir Mesylate (NFV) is a non-peptidic HIV-1 protease inhibitor (IP) which was the first IP recommended for pediatric use (>2 years-old). It exhibits pH-dependant aqueous solubility which results highly restricted at physiological pH values. The former represents a main clinical limitation due to the reduction on drug absorption along the small intestine after an oral administration, leading to unpredictable drug bioavailability. Moreover a liquid formulation of NFV is not available worldwide, preventing appropriate dose adjustment and more convenient administration. In this framework, the present investigation reports the development of a NFV highly concentrated aqueous formulation for a more appropriate management of pediatric anti-HIV therapy. The aim was to encapsulate NFV within D-alpha-tocopheryl polyethylene glycol 1000 succinate micelles to improve its aqueous solubility and its oral pharmacokinetic parameters. Results show that NFV aqueous solubility was increased up to 80.3 mg/mL. NFV-loaded micelles exhibited a hydrodynamic diameter of 5.6 nm and a spherical morphology as determined by dynamic light scattering and transmission electronic microscopy, respectively. In vitro NFV release profile demonstrated a cumulative drug release of 56% at 6 h. Finally, in vivo data showed a significant (p<0.01) increase of Area-Under-the-Curve between 0 and 24 h for NFV encapsulated in micelles in comparison with a NFV suspension prepared with glycerin 20% v/v and carboxymethylcellulose sodium 0.5% w/v, representing an increment on drug oral relative bioavailability of 1.71-fold. Thereby, this formulation represents an innovative nanotechnological platform to improve pediatric HIV pharmacotherapy.
Toxicology and carcinogenesis studies of mixtures of 3'-azido-3'-deoxythymidine (AZT), lamivudine (3TC), nevirapine (NVP), and nelfinavir mesylate (NFV) (Cas Nos. 30516-87-1, 134678-17-4, 129618-40-2, 159989-65-8) in B6C3F1 Mice (transplacental exposure studies).[Pubmed:23385634]
Natl Toxicol Program Tech Rep Ser. 2013 Jan;(569):1-212.
BACKGROUND: Antiretroviral drugs are used to treat patients positive for the human immunovirus HIV-1, and increasingly treatments include a combination of such drugs. The noninfected children of women who are pregnant and receiving such treatment may also be exposed to the drugs by transplacental exposure. We studied the long-term effects of such transplacental exposure in mice by exposing pregnant mice to combinations of four such antiretroviral drugs for seven days and then observing their pups for two years following birth. The four drugs studied were 3'-azido-3'-deoxythymidine (AZT), lamivudine (3TC), nevirapine (NVP), and Nelfinavir Mesylate (NFV). METHODS: Four different sets of exposure studies were performed: exposure to AZT; to AZT plus 3TC; to AZT, 3TC, and NVP; or to AZT, 3TC, and NFV. In each of these studies, groups of pregnant females were given one of three concentrations of the drug combinations seven times though a tube directly into their stomachs, and after birth their pups were maintained with no further exposure for two years. The offspring of another group of pregnant females not treated with the drugs served as controls. At the end of the study, tissues from more than 40 sites were examined for every animal. RESULTS: Survival of pups whose mothers were exposed to AZT or AZT plus 3TC was similar to their controls, while the survival rates for offspring of mice exposed to AZT, 3TC, and NVP or AZT, 3TC, and NFP were lower than for controls. In most cases the body weights of pups from mothers exposed were slightly less than those of the controls. There were slight increases in the incidences of thyroid gland tumors and skin tumors in the female pups of mothers exposed to AZT alone and of lung tumors in female pups of mothers exposed to AZT plus 3TC. For offspring of mothers exposed to AZT, 3TC, and NVP there were increased incidences of skin tumors in both male and female pups, and more so in the males. CONCLUSIONS: We conclude that exposure to the combination of AZT, 3TC, and NVP during pregnancy caused an increase in skin tumors in the male offspring and possibly also to the female offspring. Exposure to AZT alone during pregnancy may have been related to thyroid gland or skin tumors in female offspring, and exposure to AZT plus 3TC may have been related to lung tumors in female offspring.
Development and optimization of solid self-nanoemulsifying drug delivery system (S-SNEDDS) using Scheffe's design for improvement of oral bioavailability of nelfinavir mesylate.[Pubmed:25786731]
Drug Deliv Transl Res. 2014 Apr;4(2):171-86.
The present research was aimed at development and evaluation of self-nanoemulsifying drug delivery system (SNEDDS) for improving bioavailability of Nelfinavir Mesylate (NFV), a protease inhibitor exhibiting pH dependent solubility and variable oral bioavailability. Maisine 35-1, Cremophor RH-40, and Labrasol were identified as oil, surfactant, and co-surfactant that had best solubility for NFV. Scheffe's mixture design was used to optimize the amount of components in liquid self-nanoemulsifying drug delivery system (L-SNEDDS) by taking their amounts as independent variable, whereas globule size, drug loading, and percent transmittance were taken as dependent variable. Optimized NFV-L-SNEDDS was then adsorbed on Neusilin US2 to form solid self-nanoemulsifying drug delivery system (S-SNEDDS). NFV loaded L-SNEDDS and S-SNEDDS were characterized for various physicochemical properties, and solid-state properties were determined through differential scanning calorimetry, X-ray diffraction, and scanning electron microscopy studies. In vitro dissolution using simulated gastric fluid and simulated intestinal fluid, ex vivo drug release study, and in vivo study were performed for pure NFV and NFV-S-SNEDDS. NFV-S-SNEDDS showed more than 90 % drug release in 20 min during drug release studies irrespective of pH of the dissolution medium. In vivo study revealed significant difference between release of NFV from suspension and NFV-L-SNEDDS and NFV-S-SNEDDS when given to rabbits (p < 0.001). NFV-L-SNEDDS and NFV-S-SNEDDS were subjected to stability study as per ICH guidelines, and NFV-S-SNEDDS was found to be stable during the period of study. S-SNEDDS could serve as a potential drug delivery system for NFV.
Pharmacokinetics of ciprofloxacin in pediatric cystic fibrosis patients.[Pubmed:8787874]
Antimicrob Agents Chemother. 1996 Jan;40(1):29-34.
The pharmacokinetic characteristics of ciprofloxacin were studied in 10 children with cystic fibrosis, aged from 6 to 16 years, who had completed the standard regimen of intravenous ceftazidime and amikacin. The aim of the investigation was to derive dosing guidelines for young cystic fibrosis patients to be treated with ciprofloxacin. Each child received ciprofloxacin given as two 30-min infusions (10 mg/kg of body weight each) 12 h apart; this was followed by the administration of oral ciprofloxacin (15 mg/kg every 12 h). Blood samples were taken after both infusions and after the first oral dose. A total of 232 ciprofloxacin concentrations (203 concentrations in plasma and 29 concentrations in urine) were analyzed by use of NONMEM and a two-compartment body model with seven parameters: total body clearance (CL), volume of the central compartment (V2), volume of the peripheral compartment (V3), intercompartmental clearance, renal clearance, absorption rate constant, and bioavailability. The influences of weight (range, 18 to 42 kg) and age (range, 6 to 16 years) were investigated. CL (in liters per hour) was found to be linearly correlated with weight (typical value of CL = 8.8 + 0.396. WT, where WT is weight; (interindividual coefficient of variation, 7.8%). V2 and V3 were directly proportional to weight, with slopes of 0.7 and 1.3 liters/kg, respectively. Interindividual variabilities were calculated to be 22.6 and 14.9% for V2 and V3, respectively. No dependency of the other pharmacokinetic parameters on age or weight was seen. Because of the high correlations between age and weight, only one covariable was necessary. Weight had the strongest effect. Bioavailability (population mean) was estimated to be 61.8%, and renal clearance (population mean) was estimated to be 11.4 liters/h. The residual (intraindividual) variability was 31.9%. The protein binding was about 34%, which is similar to the results obtained for adults. In order to define the appropriate dosage regimen for children suffering from cystic fibrosis, a formula was derived so that steady-state concentrations, similar to those obtained in adults after the administration of dosages of 400 mg three times daily intravenously and 750 mg twice daily orally, could be reached. The calculated total daily dose increased with increasing body weight. Given as milligrams per kilogram of body weight, the calculated dosage regimens suggest that for younger children (weight range, 14 to 28 kg), 28 to 20 mg/kg orally twice daily should be given, and for older children (weight range, 28 to 42 kg), 20 to 15 mg/kg orally twice daily should be given. For intravenous administration, dosages of 15 to 10 mg/kg twice daily are sufficient.
Preclinical pharmacokinetics and distribution to tissue of AG1343, an inhibitor of human immunodeficiency virus type 1 protease.[Pubmed:8787890]
Antimicrob Agents Chemother. 1996 Jan;40(1):110-4.
AG1343, a potent inhibitor of human immunodeficiency virus type 1 (HIV-1) protease (Ki = 2 nM), was designed by protein structure-based drug design techniques. AG1343 has potent antiviral activity (95% effective dose = 0.04 microgram/ml) against a number of HIV-1 strains in acute and chronic models of infection. As part of its preclinical development, the oral bioavailability of AG1343 in rats, dogs, monkeys, and marmosets was determined and its tissue distribution in rats was evaluated. There were no major interspecies differences in AG1343 pharmacokinetics. Following intravenous administration, the elimination half-life of AG1343 ranged from 1 to 1.4 hr. The total volume of distribution (2 to 7 liters/kg) exceeded the volume of total body water, indicating extensive tissue distribution. Systemic clearance of AG1343 (1 to 4 liters/kg) in the different species corresponded to hepatic blood flow, suggesting possible hepatic involvement in the elimination of AG1343. Following oral administration, peak levels in plasma ranged from 0.34 microgram/ml after treatment with 10 mg/kg of body weight in the dog to 1.7 micrograms/ml after dosing with 50 mg/kg in the rat. Because of the slow absorption of AG1343, plasma concentrations of AG1343 exceeding that required for 95% inhibition of HIV-1 replication were maintained for up to 7 h after a single oral dose in all species evaluated. Average oral bioavailability of AG1343 ranged from 17% in the marmoset to 47% in the dog. Studies of distribution to tissue in the rat after oral administration of 14C-AG1343 established extensive distribution with concentrations in most tissues exceeding that found in plasma. Of particular significance were high levels of AG1343 equivalent in mesenteric lymph nodes (32.05 micrograms/g) and spleen tissue (9.33 micrograms/g). The major excretory route for AG1343 was via feces, with 100% of the dose recovered by 48 h. Results from these studies demonstrate that AG1343 is orally bioavailable and that levels in plasma in the therapeutic range are achievable and are maintained for prolonged periods in the animal models tested. On the basis of these and other findings, AG1343 was developed for further testing in human subjects.
