1-Naphthyl PP1Src family kinases inhibitor CAS# 221243-82-9 |

- Imatinib Mesylate (STI571)
Catalog No.:BCC1115
CAS No.:220127-57-1
- Dasatinib (BMS-354825)
Catalog No.:BCC1281
CAS No.:302962-49-8
- Saracatinib (AZD0530)
Catalog No.:BCC1166
CAS No.:379231-04-6
- Bosutinib (SKI-606)
Catalog No.:BCC1167
CAS No.:380843-75-4
- DPH
Catalog No.:BCC1538
CAS No.:484049-04-9
Quality Control & MSDS
Number of papers citing our products
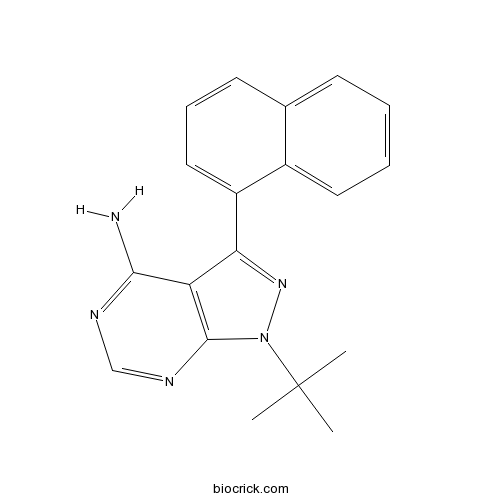
Chemical structure
3D structure
| Cas No. | 221243-82-9 | SDF | Download SDF |
| PubChem ID | 4877 | Appearance | Powder |
| Formula | C19H19N5 | M.Wt | 317.39 |
| Type of Compound | N/A | Storage | Desiccate at -20°C |
| Synonyms | 1-NA-PP 1 | ||
| Solubility | DMSO : 12.5 mg/mL (39.38 mM; Need ultrasonic) | ||
| Chemical Name | 1-tert-butyl-3-naphthalen-1-ylpyrazolo[3,4-d]pyrimidin-4-amine | ||
| SMILES | CC(C)(C)N1C2=C(C(=N1)C3=CC=CC4=CC=CC=C43)C(=NC=N2)N | ||
| Standard InChIKey | XSHQBIXMLULFEV-UHFFFAOYSA-N | ||
| Standard InChI | InChI=1S/C19H19N5/c1-19(2,3)24-18-15(17(20)21-11-22-18)16(23-24)14-10-6-8-12-7-4-5-9-13(12)14/h4-11H,1-3H3,(H2,20,21,22) | ||
| General tips | For obtaining a higher solubility , please warm the tube at 37 ℃ and shake it in the ultrasonic bath for a while.Stock solution can be stored below -20℃ for several months. We recommend that you prepare and use the solution on the same day. However, if the test schedule requires, the stock solutions can be prepared in advance, and the stock solution must be sealed and stored below -20℃. In general, the stock solution can be kept for several months. Before use, we recommend that you leave the vial at room temperature for at least an hour before opening it. |
||
| About Packaging | 1. The packaging of the product may be reversed during transportation, cause the high purity compounds to adhere to the neck or cap of the vial.Take the vail out of its packaging and shake gently until the compounds fall to the bottom of the vial. 2. For liquid products, please centrifuge at 500xg to gather the liquid to the bottom of the vial. 3. Try to avoid loss or contamination during the experiment. |
||
| Shipping Condition | Packaging according to customer requirements(5mg, 10mg, 20mg and more). Ship via FedEx, DHL, UPS, EMS or other couriers with RT, or blue ice upon request. | ||
| Description | Selective inhibitor of src family kinases v-Src and c-Fyn as well as the tyrosine kinase c-Abl. (IC50 values are 1.0, 0.6, 0.6, 18 and 22 μM for v-Src, c-Fyn, c-Abl, CDK2 and CAMK II respectively). Preferentially inhibits mutant over wild-type kinases (IC50 values are 1.5 vs 1000 nM for I338G v-src and v-src respectively). |

1-Naphthyl PP1 Dilution Calculator
1-Naphthyl PP1 Molarity Calculator
| 1 mg | 5 mg | 10 mg | 20 mg | 25 mg | |
| 1 mM | 3.1507 mL | 15.7535 mL | 31.507 mL | 63.014 mL | 78.7674 mL |
| 5 mM | 0.6301 mL | 3.1507 mL | 6.3014 mL | 12.6028 mL | 15.7535 mL |
| 10 mM | 0.3151 mL | 1.5753 mL | 3.1507 mL | 6.3014 mL | 7.8767 mL |
| 50 mM | 0.063 mL | 0.3151 mL | 0.6301 mL | 1.2603 mL | 1.5753 mL |
| 100 mM | 0.0315 mL | 0.1575 mL | 0.3151 mL | 0.6301 mL | 0.7877 mL |
| * Note: If you are in the process of experiment, it's necessary to make the dilution ratios of the samples. The dilution data above is only for reference. Normally, it's can get a better solubility within lower of Concentrations. | |||||

Calcutta University

University of Minnesota

University of Maryland School of Medicine

University of Illinois at Chicago

The Ohio State University

University of Zurich

Harvard University

Colorado State University

Auburn University

Yale University

Worcester Polytechnic Institute

Washington State University

Stanford University

University of Leipzig

Universidade da Beira Interior

The Institute of Cancer Research

Heidelberg University

University of Amsterdam

University of Auckland

TsingHua University

The University of Michigan

Miami University

DRURY University

Jilin University

Fudan University

Wuhan University

Sun Yat-sen University

Universite de Paris

Deemed University

Auckland University

The University of Tokyo

Korea University
Selective inhibitor of src family kinases v-Src and c-Fyn as well as the tyrosine kinase c-Abl. (IC50 values are 1.0, 0.6, 0.6, 18 and 22 μM for v-Src, c-Fyn, c-Abl, CDK2 and CAMK II respectively). Preferentially inhibits mutant over wild-type kinases (IC
- Z-Lys(Boc)-OH.DCHA
Catalog No.:BCC2591
CAS No.:2212-76-2
- Z-Lys-OH
Catalog No.:BCC2764
CAS No.:2212-75-1
- Glyoxalase I inhibitor
Catalog No.:BCC1598
CAS No.:221174-33-0
- Erysubin B
Catalog No.:BCN4947
CAS No.:221150-19-2
- Erysubin A
Catalog No.:BCN4946
CAS No.:221150-18-1
- 1alpha,4beta,10beta-Trihydroxyguaia-2,11(13)-dien-12,6alpha-olide
Catalog No.:BCN7483
CAS No.:221148-94-3
- 1''-Methoxyerythrinin C
Catalog No.:BCN3966
CAS No.:221002-11-5
- Guaiacol glycidyl ether
Catalog No.:BCC8992
CAS No.:2210-74-4
- Lumiracoxib
Catalog No.:BCC4440
CAS No.:220991-20-8
- Antalarmin hydrochloride
Catalog No.:BCC7480
CAS No.:220953-69-5
- 13-O-Deacetyltaxumairol Z
Catalog No.:BCN4945
CAS No.:220935-39-7
- 13-O-Cinnamoylbaccatin III
Catalog No.:BCN7344
CAS No.:220932-65-0
- 1-NM-PP1
Catalog No.:BCC4306
CAS No.:221244-14-0
- 7,4'-Di-O-methylapigenin 5-O-xylosylglucoside
Catalog No.:BCN1484
CAS No.:221257-06-3
- Lethedoside A
Catalog No.:BCN4948
CAS No.:221289-20-9
- Lethedioside A
Catalog No.:BCN5046
CAS No.:221289-31-2
- Leptocarpinine
Catalog No.:BCN3730
CAS No.:221347-12-2
- Podocarpusflavone A
Catalog No.:BCN5047
CAS No.:22136-74-9
- Pinosylvin
Catalog No.:BCN5048
CAS No.:22139-77-1
- Cytochalasin D
Catalog No.:BCN5049
CAS No.:22144-77-0
- Fischeria A
Catalog No.:BCN3779
CAS No.:221456-63-9
- 2-Oxokolavelool
Catalog No.:BCN4672
CAS No.:221466-41-7
- 2 beta-Hydroxykolavelool
Catalog No.:BCN4671
CAS No.:221466-42-8
- 11-Hydroxytabersonine
Catalog No.:BCN5050
CAS No.:22149-28-6
A single amino-acid change in ERK1/2 makes the enzyme susceptible to PP1 derivatives.[Pubmed:16431218]
Biochem Biophys Res Commun. 2006 Mar 3;341(1):261-5.
We generated extracellular signal-regulated kinase 1/2 (ERK1/2) mutants by introducing a single amino-acid substitution in subdomain V of the catalytic domain and then examined the susceptibility of these mutants to PP1 derivatives originally designed as Src inhibitors. Substituting smaller amino acids (alanine [Ala (A)] or glycine [Gly (G)]) for glutamine [Gln (Q)] in subdomain V drastically increased the susceptibility of ERK1/2 to 1-Naphthyl PP1 (1NA-PP1). Wild-type ERK1/2 was resistant to 1NA-PP1 inhibition. ERK1(Q122A) and ERK2(Q103A) were inhibited by 1NA-PP1 at IC(50) values of 1.7 +/- 0.13 and 2.1 +/- 0.18 microM, respectively. ERK1(Q122G) and ERK2(Q103G) were inhibited by 1NA-PP1 with IC(50) values of 3.6 +/- 0.26 and 18 +/- 2.2 microM, respectively. Other derivatives of PP1 (1-naphthylmethyl PP1 and 2-naphthylmethyl PP1) did not significantly inhibit ERK1/2 and its various mutants. In addition, these ERK1/2 mutants were activated by TPA when they were expressed in mammalian cells. These results suggest that the Gln residue of subdomain V is important in determining the susceptibility of ERK1/2 to 1NA-PP1 without significant changes in their enzymatic characteristics.
Generation and characterization of ATP analog-specific protein kinase Cdelta.[Pubmed:25505183]
J Biol Chem. 2015 Jan 23;290(4):1936-51.
To better study the role of PKCdelta in normal function and disease, we developed an ATP analog-specific (AS) PKCdelta that is sensitive to specific kinase inhibitors and can be used to identify PKCdelta substrates. AS PKCdelta showed nearly 200 times higher affinity (Km) and 150 times higher efficiency (kcat/Km) than wild type (WT) PKCdelta toward N(6)-(benzyl)-ATP. AS PKCdelta was uniquely inhibited by 1-(tert-butyl)-3-(1-naphthyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (1NA-PP1) and 1-(tert-butyl)-3-(2-methylbenzyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (2MB-PP1) but not by other 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP1) analogs tested, whereas WT PKCdelta was insensitive to all PP1 analogs. To understand the mechanisms for specificity and affinity of these analogs, we created in silico WT and AS PKCdelta homology models based on the crystal structure of PKCiota. N(6)-(Benzyl)-ATP and ATP showed similar positioning within the purine binding pocket of AS PKCdelta, whereas N(6)-(benzyl)-ATP was displaced from the pocket of WT PKCdelta and was unable to interact with the glycine-rich loop that is required for phosphoryl transfer. The adenine rings of 1NA-PP1 and 2MB-PP1 matched the adenine ring of ATP when docked in AS PKCdelta, and this interaction prevented the potential interaction of ATP with Lys-378, Glu-428, Leu-430, and Phe-633 residues. 1NA-PP1 failed to effectively dock within WT PKCdelta. Other PP1 analogs failed to interact with either AS PKCdelta or WT PKCdelta. These results provide a structural basis for the ability of AS PKCdelta to efficiently and specifically utilize N(6)-(benzyl)-ATP as a phosphate donor and for its selective inhibition by 1NA-PP1 and 2MB-PP1. Such homology modeling could prove useful in designing molecules to target PKCdelta and other kinases to understand their function in cell signaling and to identify unique substrates.
Biochemical measurements on single erythroid progenitor cells shed light on the combinatorial regulation of red blood cell production.[Pubmed:23168618]
Mol Biosyst. 2013 Feb 2;9(2):234-45.
Adult bone marrow (BM) erythrocyte colony-forming units (CFU-Es) are important cellular targets for the treatment of anemia and also for the manufacture of red blood cells (RBCs) ex vivo. We obtained quantitative biochemical measurements from single and small numbers of CFU-Es by isolating and analyzing c-Kit(+)CD71(high)Ter119(-) cells from adult mouse BM and this allowed us to identify two mechanisms that can be manipulated to increase RBC production. As expected, maximum RBC output was obtained when CFU-Es were stimulated with a combination of Stem Cell Factor (SCF) and Erythropoietin (EPO) mainly because SCF supports a transient CFU-E expansion and EPO promotes the survival and terminal differentiation of erythroid progenitors. However, we found that one of the main factors limiting the output in RBCs was that EPO induces a downregulation of c-Kit expression which limits the transient expansion of CFU-Es. In the presence of SCF, the EPO-mediated downregulation of c-Kit on CFU-Es is delayed but still significant. Moreover, treatment of CFU-Es with 1-Naphthyl PP1 could partially inhibit the downregulation of c-Kit induced by EPO, suggesting that this process is dependent on a Src family kinase, v-Src and/or c-Fyn. We also found that CFU-E survival and proliferation was dependent on the level of time-integrated extracellular-regulated kinase (ERK) activation in these cells, all of which could be significantly increased when SCF and EPO were combined with mouse fetal liver-derived factors. Taken together, these results suggest two novel molecular strategies to increase RBC production and regeneration.
Aurora A is involved in central spindle assembly through phosphorylation of Ser 19 in P150Glued.[Pubmed:23547029]
J Cell Biol. 2013 Apr 1;201(1):65-79.
Knowledge of Aurora A kinase functions is limited to premetaphase events, particularly centrosome maturation, G2/M transition, and mitotic spindle assembly. The involvement of Aurora A in events after metaphase has only been suggested because appropriate experiments are technically difficult. We report here the design of the first human Aurora A kinase (as-AurA) engineered by chemical genetics techniques. This kinase is fully functional biochemically and in cells, and is rapidly and specifically inhibited by the ATP analogue 1-Naphthyl-PP1 (1-Na-PP1). By treating cells exclusively expressing the as-AurA with 1-Na-PP1, we discovered that Aurora A is required for central spindle assembly in anaphase through phosphorylation of Ser 19 of P150Glued. This paper thus describes a new Aurora A function that takes place after the metaphase-to-anaphase transition and a new powerful tool to search for and study new Aurora A functions.
New pyrazolopyrimidine inhibitors of protein kinase d as potent anticancer agents for prostate cancer cells.[Pubmed:24086585]
PLoS One. 2013 Sep 23;8(9):e75601.
The emergence of protein kinase D (PKD) as a potential therapeutic target for several diseases including cancer has triggered the search for potent, selective, and cell-permeable small molecule inhibitors. In this study, we describe the identification, in vitro characterization, structure-activity analysis, and biological evaluation of a novel PKD inhibitory scaffold exemplified by 1-Naphthyl PP1 (1-NA-PP1). 1-NA-PP1 and IKK-16 were identified as pan-PKD inhibitors in a small-scale targeted kinase inhibitor library assay. Both screening hits inhibited PKD isoforms at about 100 nM and were ATP-competitive inhibitors. Analysis of several related kinases indicated that 1-NA-PP1 was highly selective for PKD as compared to IKK-16. SAR analysis showed that 1-NA-PP1 was considerably more potent and showed distinct substituent effects at the pyrazolopyrimidine core. 1-NA-PP1 was cell-active, and potently blocked prostate cancer cell proliferation by inducing G2/M arrest. It also potently blocked the migration and invasion of prostate cancer cells, demonstrating promising anticancer activities on multiple fronts. Overexpression of PKD1 or PKD3 almost completely reversed the growth arrest and the inhibition of tumor cell invasion caused by 1-NA-PP1, indicating that its anti-proliferative and anti-invasive activities were mediated through the inhibition of PKD. Interestingly, a 12-fold increase in sensitivity to 1-NA-PP1 could be achieved by engineering a gatekeeper mutation in the active site of PKD1, suggesting that 1-NA-PP1 could be paired with the analog-sensitive PKD1(M659G) for dissecting PKD-specific functions and signaling pathways in various biological systems.
A chemical switch for inhibitor-sensitive alleles of any protein kinase.[Pubmed:11014197]
Nature. 2000 Sep 21;407(6802):395-401.
Protein kinases have proved to be largely resistant to the design of highly specific inhibitors, even with the aid of combinatorial chemistry. The lack of these reagents has complicated efforts to assign specific signalling roles to individual kinases. Here we describe a chemical genetic strategy for sensitizing protein kinases to cell-permeable molecules that do not inhibit wild-type kinases. From two inhibitor scaffolds, we have identified potent and selective inhibitors for sensitized kinases from five distinct subfamilies. Tyrosine and serine/threonine kinases are equally amenable to this approach. We have analysed a budding yeast strain carrying an inhibitor-sensitive form of the cyclin-dependent kinase Cdc28 (CDK1) in place of the wild-type protein. Specific inhibition of Cdc28 in vivo caused a pre-mitotic cell-cycle arrest that is distinct from the G1 arrest typically observed in temperature-sensitive cdc28 mutants. The mutation that confers inhibitor-sensitivity is easily identifiable from primary sequence alignments. Thus, this approach can be used to systematically generate conditional alleles of protein kinases, allowing for rapid functional characterization of members of this important gene family.
